19/06 – Wereld Sikkelcel Dag: 1ste genetische ziekte in Brussel
Wist u dat in Brussel de sikkelcelziekte de eerste genetische ziekte was? Het treft 1 pasgeborene op 1900 geboortes. Met pijnlijk en met zware gevolgen, deze chronische ziekte is een erfelijke aandoening van het hemoglobine eiwit in de rode bloedcellen. Deze ziekte vereist een globale en multidisciplinaire aanpak met een verschillende diensten en zorgverleners.
Ter gelegenheid van de Wereld Sikkelcel Dag deze 19 juni, wijden wij hieraan een column in onze blog. Een interview met prof. Alina Ferster, hoofd van de dienst hematologie-oncologie van het Universitair Kinderziekenhuis Koningin Fabiola.
“Kinderen met deze ziekte leiden aan bloedarmoede. Ze hebben pijnlijke aanvallen, herhaald en onvoorspelbaar, gerelateerd aan de obstructie van de kleine bloedvaten, en kan mogelijk ernstige infecties ontwikkelen”, legt Dr. Alina Ferster uit. “De meervoudige orgaancomplicaties die meestal de hersenen, het hart, de longen, de nieren, de botten en ogen raken, kunnen zich mettertijd voordoen. Deze complicaties komen frequent voor in de volwassenheid. ”
Sikkelcelpatiënten worden op een specifieke manier ondersteund door het UKZKF. “Momenteel worden meer dan 200 kinderen en adolescenten opgevolgd door artsen en verplegers van de afdeling hematologie”, verduidelijkt Prof. Ferster. De ondersteuning is gebaseerd op een geïntegreerde, globale aanpak gebaseerd op de volgende punten:
- Informatie aan ouders van pasgeborenen over de neonatale screening, georganiseerd op een ongerichte manier (voor alle kinderen, ongeacht hun herkomst) in de Brusselse regio.
- Informatie over patiënten, hun ouders, hun naaste omgeving, maar ook de psychologische hulp alsook de sociale ondersteuning van de patiënten en hun gezin.
- Infectiepreventie door middel van vaccinatie, in het bijzonder voor pneumokokken, en door het toedienen van antibiotica tijdens de eerste levensjaren.
- Screening en pre-symptomatische behandeling van meervoudige complicaties door de occlusie van bloedvaten(hersenen, ogen,…), alsook het toedienen van een behandeling met hydroxyurea voor de meest symptomatische patiënten. Dit middel is tot dusver het enige dat zijn belang bewezen heeft door het reduceren van het aantal sterftes ten gevolge van sikkelcelziekte.
- De optimale behandeling van acute en chronische complicaties, dankzij de medewerking en betrokkenheid van de hulpdiensten, de intensive care, opname units, evenals de medewerking van alle artsen van
 verscheidene medische en chirurgische specialisaties in het UKZKF.
verscheidene medische en chirurgische specialisaties in het UKZKF.
Beenmergtransplantatie en behandelingen die de ziekte wijzigen
Hydroxyurea, een chronische transfusie en een beenmergtransplantatie zijn momenteel de enige beschikbare benaderingen die de symptomen van de ziekte veranderen. De indicatie om deze behandelingen te beginnen wordt regelmatig getoetst door het medische team.
“De beenmergtransplantatie blijft momenteel de enige behandeling die de sikkelcelziekte kan genezen. Deze behandeling is complex en kan leiden tot verschillende complicaties. Het is geschikt voor kinderen met een compatibele donor binnen de familie. In het UKZKF is ons transplantatieprogramma binnen de sikkelcelziekte gestart in 1989 en heeft dusver 65 kinderen bevoordeeld. De huidige resultaten zijn uitstekend, ” aldus Dr. Ferster. Innovatieve benaderingen bij transplantatie zijn in ontwikkeling, waaronder gentherapie. Verschillende veelbelovende geneesmiddelen zullen binnenkort beschikbaar zijn binnen het kader van klinische studies.
Evolutie van de ondersteuning
“Sikkelcelziekte mag niet langer worden beschouwd als een ziekte van hemoglobine. Het moet gezien en begrepen worden als een ernstige, systemische ziekte die invloed kan hebben op veel organen en waarvan de dreigende acute complicaties een onomkeerbare handicap kan veroorzaken,” zei Prof. Fester. In de afgelopen jaren was optimalisatie in transfusiegeneeskunde essentieel voor de kwaliteitszorg, want het was nodig voor de behandeling van sommige kinderen en de behandeling van pijn met behulp van specifiek opgeleide professionals. “Vandaag de dag bereikt de overgrote meerderheid van de getroffen kinderen de volwassenheid. Een globale aanpak van de ziekte kan de mortaliteit en morbiditeit bij kinderen aanzienlijk verminderen.” Het UKZKF werkt ook nauw samen met de hematologie-oncologie dienst van UVC Brugmann, doordat op volwassen leeftijd sikkelcelanemie nieuwe uitdagingen definieert voor de artsen en de ziekenhuizen.
:: 1 pasgeborene op 1900::
De homozygote sikkelcelziekte is de meest voorkomende genetische ziekte van hemoglobine ter wereld. Het treft ongeveer 1% van de geboorten in sommige landen in Centraal-Afrika, maar is ook niet ongewoon in Europa. Ze werd doorheen de migratie de meest voorkomende genetische ziekte in de Brusselse regio, die 1 pasgeborene van de 1900 treft.
:: Een genetische ziekte ::
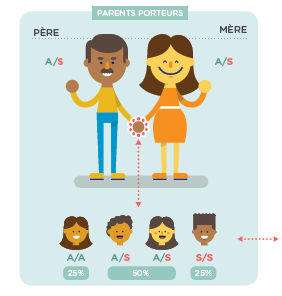
Sikkelcelziekte is een erfelijke ziekte die autosomaal dominant wordt overgedragen: dragers van de genetische mutatie (“gezonde dragers”) vertonen geen symptomen. Om ziek te zijn moet het kind het zieke gen van elk van zijn ouders hebben geërfd (er is een kans van 25% wanneer beide ouders drager zijn van de mutatie)

:: Een specifieke ondersteuning ::
De ondersteuning van kinderen met sikkelcelziekte wordt verzekerd door acht artsen van de dienst hematologie van het UKZKF: Dr. Ferster, Dr. Le, Dr. Heijmans, Dr. Huybrecht, Dr. Azzi, Dr. Devalck, Dr. Dedeken en Dr. Diallo. De voorlichting en educatie van de patiënten en hun families (Hoe risicovolle situaties opsporen die een dringende hospitalisatie vereisen?) worden verzekerd door een verwijzende, halftijdse verpleegster.
Een psychologe en een maatschappelijk werkster zijn verbonden aan de dienst hematologie-oncologie om jonge patiënten en hun naasten te ondersteunen.
Er zijn doorschakelingen opgezet met de eerste hulp, intensive care, one-day units, verschillende specialisten van het UKZKF en hematologen van het UVC Brugmann.
Bron:
Extracten van Osiris News nr. 21, december 2010 – februari 2011
Update 19/06/2017





